How can we document software and computational analyses in such a way that others can convince themselves of their validity, and build on them for their own work? The question has been around for many years, and a number of attempts have been made to provide partial answers. This post provides a brief review and describes my own tentative answer, inviting you to play with it.
Thirty years after my first contact with computational (ir)reproducibility, I am happy to note that many things have improved. Reproducibility, computational and otherwise, is increasingly recognized as an important aspect of scientific quality control, and mostly considered worth striving for. However, I also note that more and more people, including reproducibility activists, have lost contact with the day-to-day reality in which reproducibility matters. Reproducibility is becoming an item on a checklist, and its precise incarnation the subject of political bickering aimed at making it easy to check off that item. So let's take a look at why computational reproducibility matters for researchers.
At the recent SciCodes Symposium, I brought up the question of reviewing research software during the panel discussion. One panelist then raised the question of why we should review research software. I found this question surprising at first, but I do agree that it deserves an answer. Here is mine.
In my last post, I have discussed the two main types of scientific models: empirical models, also called descriptive models, and explanatory models. I have also emphasized the crucial role of equations and specifications in the formulation of explanatory models. But my description of scientific models in that post left aside a very important aspect: on a more fundamental level, all models are stories.
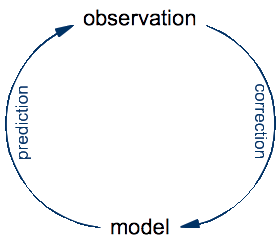
It is often said that science rests on two pillars, experiment and theory. Which has lead some to propose one or two additional pillars for the computing age: simulation and data analysis. However, the real two pillars of science are observations and models. Observations are the input to science, in the form of numerous but incomplete and imperfect views on reality. Models are the inner state of science. They represent our current understanding of reality, which is necessarily incomplete and imperfect, but understandable and applicable. Simulation and data analysis are tools for interfacing and thus comparing observations and models. They don't add new pillars, but transforms both of them. In the following, I will look at how computing is transforming scientific models.
One question I have been thinking about in the context of reproducible research is this: Why is all stable software technology old, and all recent technology fragile? Why is it easier to run 40-year-old Fortran code than ten-year-old Python code? A hypothesis that comes to mind immediately is growing code complexity, but I'd expect this to be an amplifier rather than a cause. In this pose, I will look at another candidate: the dominance of Open Source communities in the development of scientific software.
It's the season when everyone writes about the past year, or even the past decade for a year number ending in 9. I'll make a modest contribution by summarizing my experience with Pharo after one year of using it for projects of my own.
Regular readers of this blog may have noticed that I am not very happy with today's state of computational notebooks, such as they were pioneered by Mathematica and popularized by more recent free incarnations such as Jupyter, R markdown, or Emacs/OrgMode. In this post and the accompanying screencast (my first one!), I will explain what I dislike about today's notebooks, and how I think we can do better.
One of the more interesting things I have been playing with recently is Pharo, a modern descendent of Smalltalk. This is a summary of my first impressions after using it on a small (and unfinished) project, for which it might actually turn out to be very helpful.
There is an important and ubiquitous process in scientific research that scientists never seem to talk about. There isn't even a word for it, as far as I now, so I'll introduce my own: I'll call it knowledge distillation.
In today's scientific practice, there are two main variants of this process, one for individual research studies and one for managing the collective knowledge of a discipline. I'll briefly present both of them, before coming to the main point of this post, which is the integration of digital knowledge, and in particular software, into the knowledge distillation process.
Since the dawn of computer programming, software developers have been aware of the rapidly growing complexity of code as its size increases. Keeping in mind all the details in a few hundred lines of code is not trivial, and understanding someone else's code is even more difficult because many higher-level decisions about algorithms and data structures are not visible unless the authors have carefully documented them and keep those comments up to date.
My most recent paper submission (preprint available) is about improving the verifiability of computer-aided research, and contains many references to the related subject of reproducibility. A reviewer asked the same question about all these references: isn't this the same as for experiments done with lab equipment? Is software worse? I think the answers are of general interest, so here they are.
It is widely recognized by now that software is an important ingredient to modern scientific research. If we want to check that published results are valid, and if we want to build on our colleagues' published work, we must have access to the software and data that were used in the computations. The latest high-impact statement along these lines is a Nature editorial that argues that with any manuscript submission, authors should also submit the data and the software for review. I am all for that, and I hope that more journals will follow.
Think of all the things you hate about using computers in doing research. Software installation. Getting your colleagues' scripts to work on your machine. System updates that break your computational code. The multitude of file formats and the eternal need for conversion. That great library that's unfortunately written in the wrong language for you. Dependency and provenance tracking. Irreproducible computations. They all have something in common: they are consequences of the difficulty of composing digital information. In the following, I will explain the root causes of these problem. That won't make them go away, but understanding the issues will perhaps help you to deal with them more efficiently, and to avoid them as much as possible in the future.
A recurrent theme in computational science (and elsewhere) is the need to combine machine-readable information (which in the following I will call "facts" for simplicity) with a narrative for the benefit of human readers. The most obvious situation is a scientific publication, which is essentially a narrative explaining the context and motivation for a study, the work that was undertaken, the results that were observed, and conclusions drawn from these results. For a scientific study that made use of computation (which is almost all of today's research work), the narrative refers to various computational facts, in particular machine-readable input data, program code, and computed results.
Like all information with a complex structure, scientific knowledge evolves over time. New ideas turn into validated models, and are ultimately integrated into a coherent body of knowledge defined by the concensus of a scientific community. In this essay, I explore how this process is affected by the ever increasing use of computers in scientific research. More precisely, I look at "digital scientific knowledge", by which I mean scientific knowledge that is processed using computers. This includes both software and digital datasets. For simplicity, I will concentrate on software, but much of the reasoning applies to datasets as well, if only because the precise meaning of non-trivial datasets is often defined by the software that treats them.
Yesterday I participated (as a visitor) in the kickoff meeting for OpenDreamKit, where one recurrent topic of discussion was notebooks, both Jupyter and Sage, including the question if they could be brought together. This reminded me of a recent blog post by Kirill Pomogajko entitled "Why I don't like Jupyter". And it reminded me of my own long-term project of integrating Jupyter with my ActivePapers system for reproducible research. That's three reasons for writing down my thoughts about notebooks and their role(s) in computational research, so here we go.
One key observation is in Gaël Varoquaux's comment on Kirill's blog post: using Jupyter for doing science creates a lock-in, because all collaborators on a project must agree on using Jupyter. There is no other tool that can be used productively for working with notebooks. It's a case of "wordization": digital content is taken hostage by a tool that defines a storage format for its own convenience without much consideration for other tools, be they competing or complementary. Wordization not only restricts the users' freedom to work with their data, but also creates headaches for the future. A data format defined by a tool can easily become unusable as the tool evolves and introduces incompatibilities, or of course if it disappears. In the case of Jupyter, its developers have always provided upgrade paths for notebooks between versions, but at some time this is bound to create trouble. Bugs are a fact of life, and I don't expect that the version-2-compatibility-feature will get much testing in Jupyter version 23. To make it worse, a Jupyter notebook can depend on third-party code that implements embedded widgets. This is one of the reasons why I don't use Jupyter for my research, although I am a big fan of using it for teaching. The other reason is that I cannot usefully link a notebook to other relevant information, such as code and data dependencies. Jupyter doesn't provide any functionality for this, and they are hard to implement externally exactly because of wordization.
Wordization is often associated with evil intentions of market dominance, as they are regularly assumed for a company like Microsoft. But I believe that the fundamental cause is the obsession with tools over content that has driven the computing industry for many years. The tool aspects of a piece of software, such as its feature list and its user interface, are immediately visible. On the contrary, its data model attracts attention only by a few specialists, if at all. Users feel the consequences of bad (or absent) data model design through the symptoms of wordization, in particular lock-in, but rarely understand where it comes from. Interestingly, this problem was also mentioned yesterday at the OpenDreamKit meeting, by Michael Kohlhase who discussed the digital representation of mathematical knowledge and the difficulty of exchanging it between different software tools. I have written earlier about another aspect, the representation of scientific models in computational science, which illustrates the extreme case of tools having absorbed scientific content to the point that its users don't even realize that something is missing.
Back to notebooks. Let's forget about tools for the moment and consider the question of what a notebook actually is, as a digital document. I think that notebooks are trying to be two different things, and that many of the problems we have with them come from this ambiguity. One role of notebooks is the documentation of computational work as a narrative with direct access to the data. This is why people publish notebooks. The other role is as a protocol of interactive explorative work, i.e. the computational scientist's equivalent of a lab notebook. The two roles are not completely unrelated, but they still significatively different.
To see the difference, look at how experimental scientists worked in the good old days of pencil, paper, and the printing press. As experiments were done, all the relevant information (preparation, results, …) was written down, immediately, with a time stamp, in the lab notebook. Like a bank ledger, a lab notebook is an immutable protocol of what happened. You don't go back and change earlier entries, that would even be considered fraud. You just add information at the end. Of course, the resulting protocol is not a good way to communicate one's findings. Therefore they are distilled and written up in a separate narrative, which surrounds a description of the work and its most important results by a motivating introduction and summarizing conclusions. This is the classic scientific article.
Today's computational notebooks are trying to be both protocol and narrative, and pretend that there is a fluent transition between them. One unfortunate consequence is that computational protocols disappear as they are edited to become narratives. This could be alleviated by keeping notebooks under version control, but I have yet to see good versioning support in any notebook-type tool. But, fundamentally, today's notebook tools don't encourage keeping a protocol. They encourage frequent changes to the code and the results, keeping only the latest version. As editors for narratives, notebook tools are also far from ideal because they encourage interactive execution of small code snippets, making it easy to lose track of what was actually executed and in what order. In Jupyter, the only way to ensure a coherent narrative is to (1) restart the kernel and (2) re-execute all cells. There is not even a single menu entry for this operation. Actually, I wonder how many Jupyter users are aware that they must restart the kernel before re-executing all the cells if they want to ensure reproducibility.
With all that said, here is my current idea of what a notebook should look like at the bit level. A notebook data model should have two distinct entries, one for a protocol and one for a narrative. The protocol entry is a sequence of code cells and results, as they were executed since the start of the computation (for Jupyter, that means the last kernel restart). The narrative is a user-edited sequence of code cells, documentation cells, and results. The actual cell contents could well be shared between the two views: store each cell with a unique ID, and make the protocol and the narrative simple lists of IDs. The representation of code and documentation cells in such a data model is straightforward, though there's a huge potential for bikeshedding in defining the details. The representation of results is much more difficult if you want to support more than plain text output. In the long run, it will be inevitable to define clear data models for every type of display widget, which is a lot of work.
From the tool point of view, the current Jupyter interface could be complemented by a non-editable protocol view. I'd also like to see a single command (menu/keyboard) for the "clean slate" operation: save the current state as a snapshot (or commit it directly to version control), restart the kernel, and re-initialize the protocol to an empty list. But what really matters to me is the data model. Contrary to the current one implemented in Jupyter, the one outlined above could be integrated into workflow management and archivation tools, such as my own ActivePapers. We'd probably see an Emacs mode for working with it as well. Plus pretty-printing tools, analysis tools, etc. We'd see an ecosystem of tools working with notebooks. A Dream of Openness.
SciPy 2015 is over, meaning that many non-participants like myself are now busy catching up with what happened by watching the videos. Today's dose for me was Jake VanderPlas' keynote entitled "State of the Tools". It's about the history, current state, and potential future of what is now generally known as the Scientific Python ecosystem: the large number of libraries and tools written in or for Python that scientists from many disciplines use to get their day-to-day computational work done.
History is done, the present status is a fact, but the future is open to both speculation and planning, so that's what I find most interesting in Jake's keynote. What struck me is that everything he discussed was about paying back technical debt: refactoring the core libraries, fixing compatibility problems, removing technical obstacles to installation and use of various tools. In fact, 20 years after Python showed up in scientific computing, the ecoystem is in a state that is typical for software projects of that age: a bit of a mess. The future work outlined by Jake would help to make it less of a mess, and I hope that something like this will actually happen. The big question mark for me is how this can be funded, given that it is "only" maintenance work, producing nothing fundamentally new. Fortunately there are people much better than me at thinking about funding, for example everyone involved in the NumFOCUS foundation.
Jake's approach to outlining the future is basically "how can we fix known problems and introduce some obvious improvements" (but please do watch the video to get the full story!). What I'd like to present here is an alternate approach: imagine an ideal scientific computing environment in 2015, and try to approximate it by an evolution of the current SciPy ecosystem while retaining a sane level of backwards compatibility. Think of it as the equivalent of Python 3 at the level of the core of the scientific ecosystem.
One aspect that has changed quite a bit over 20 years is the interaction between Python and low-level code. Back then, Python had an excellent C interface, which also worked well for Fortran 77 code, and the ease of wrapping C and Fortran libraries was one of the major reasons for Python's success in scientific computing. We have seen a few generations of wrapper code generators, starting with SWIG, and the idea of a hybrid language called Pyrex that was the ancestor of today's Cython. LLVM has been a major game changer, because it permits low-level code to be generated and compiled on-the-fly, without explicitly generating wrappers and compiling code. While wrapping C/C++/Fortran libraries still remains important, the equally important task of writing low-level code for performance can be handled much better with such tools. Numba is perhaps the best-known LLVM-based code generator in the Python world, providing JIT compilation for a language that is very similar to a subset of Python. But Numba is also an example of the mindset that has led to the current mess: take the existing ecosystem as given, and add a piece to it that solves a specific problem.
So how would one approach the high-/low-level interface today, having gained experience with LLVM and PyPy? Some claim that the distinction doesn't make sense any more. The authors of the Julia language, for example, claim that it "avoids the two-language problem". However, as I have pointed out on this blog, Julia is fundamentally a performance-oriented low-level language, in spite of having two features, interactivity and automatic memory management, that are traditionally associated with high-level languages. By the way, I don't believe the idea of a both-high-and-low-level language is worth pursuing for scientific computing. The closest realization of that idea is Common Lisp, which is as high-level as Python, perhaps more so, and also as low-level as Julia, but at the cost of being a very complex language with a very steep learning curve, especially for mastering the low-level aspects. Having two clearly distinct language levels makes it possible to keep both of them manageable, and the separation line serves as a clear warning sign to scientists, who should not attempt to cross it without first acquiring some serious knowledge about software development.
The model to follow, in my opinion, is the one of Lush and Terra. They embed a low-level language into a high-level language in such a way that the low-level code is a data structure at the high level. You can use literals for this data structure and get the equivalent of Numba. But you can also write code generators that specialize low-level code for a given problem. Specialization allows both optimization and simplification, both of which are desirable. The low-level language would have arrays as a primitive data structure, and both NumPy and Pandas, or evolutions such as xray, would become shallow Python APIs to such low-level array functionality. I think this is much more powerful than today's Numba building on NumPy. Moreover, wrapper generators become simple plain Python code, making the construction of interfaces to complex libraries (think of h5py) much easier than it is today. Think of it as ctypes on steroids. For more examples of what one could do with such a system, look at metaprogramming in Julia, which is exactly the same idea.
Another aspect that Jake talks about in some detail is visualization. There again, two decades of code written by people scratching their own itches has led to a mess of different libraries with a lot of overlap and no clear distinctive features. For cleaning it up, I propose the same approach: what are the needs and the available technologies for scientific visualization in 2015? We clearly want to profit from all the Web-based technologies, both for portability (think of mobile platforms) and for integration with Jupyter notebooks. But we also need to be able to integrate visualization into GUI applications. From the API point of view, we need something simple for simple plots (Toyplot looks promising), but also more sophisticad APIs for high-volume data visualization. The main barrier to overcome, in my opinion, is the current dominance of Matplotlib, which isn't particularly good in any of the categories I have outlined. Personally, I don't believe that any evolution of Matplotlib can lead to something pleasant to use, but I'd of course be happy to be proven wrong.
Perhaps the nastiest problem that Jake addresses is packaging. He seems to believe that conda is the solution, but I don't quite agree with that. Unless I missed some recent evolutions, a Python package prepared for installation through conda can only be used easily with a Python distribution built on conda as well. And that means Anaconda, because it's the only one. Since Anaconda is not Open Source, there is no way one can build a Python installation from scratch using conda. Of course, Anaconda is perfectly fine for many users. But if you need something that Anaconda does not provide, you may not be able to add it yourself. On the Mac, for example, I cannot compile C extensions compatible with Anaconda, because Mac Anaconda is built for compatibility with ancient OSX versions that are not supported by a standard XCode installation. Presumably that can be fixed, but I suspect that would be a major headache. And then, how about platforms unsupported by Anaconda?
Unfortunately I will have to leave this at the rant level, because I have no better proposition to make. Packaging has always been a mess, and will likely remain a mess, because the underlying platforms on which Python builds are already a mess. Unfortunately, it's becoming more and more of a problem as scientific Python packages grow in size and features. It's gotten to the point where I am not motivated to figure out how to install the latest version of nMOLDYN on my Mac, although I am a co-author of that program. The previous version is good enough for my own needs, and much simpler to install though already a bit tricky. That's how you get to love the command line… in 2015.
Three years ago, I first looked at the then-very-new language Julia. Back then, I concluded that there were many interesting features, but also regretted too much bad Matlab influence in the array handling.
A hands-on Julia tutorial in my neighborhood was a good occasion to take another look at this language, which has evolved quite a bit since 2012, and continues to evolve rapidly. The tutorial taught by David Sanders was an excellent introduction, and his notebooks should even be good for self-teaching. If you already have some experience in computational science, and are interested in trying Julia out on small practical applications, have a look at them.
The good news is that Julia has much improved over the years, not only by being more complete (in particular in terms of libraries), but also through changes in the language itself. More changes are about to happen with version~0.4 which is currently under development. The changes being discussed include the array behavior that I criticized three years ago. It's good to see references to APL in this discussion. I still believe that when it comes to arrays, APL and its successors are an excellent reference. It's also good to see that the Julia developers take the time to improve their language, rather than rushing towards a 1.0 release.
Due to David's tutorial, this time my contact with Julia was much more practical, working on realistic problems. This was a good occasion to appreciate many nice features of the language. Julia has taken many good features from both Lisp and APL, and combined them seamlessly into a language that, in spite of some warts, is overall a pleasure to use. A major aspect of Julia's Lisp heritage is the built-in metaprogramming support. Metaprogramming has always been difficult to grasp, which was clear as well during the tutorial. It isn't obvious at all what kind of problem it helps to solve. But everyone who has used a language with good metaprogramming support doesn't want to go back.
A distinctive feature of Julia is that it occupies a corner of the programming language universe that was almost empty until now. In scientific computing, we have traditionally had two major categories of languages. "Low-level" languages such as Fortran, C, and C++, are close to the machine level: data types reflect those directly handled by today's processors, memory management is explicit and thus left to the programmer. "High-level" languages such as Python or Mathematica present a more abstract view of computing in which resources are managed automatically and the data types and their operations are as close as possible to the mathematical concepts of arithmetic. High-level languages are typically interpreted or JIT-compiled, whereas low-level languages require an explicit compilation step, but this is not so much a feature of the language as of their age and implementation.
Julia is resolutely modern in opting for modern code transformation techniques, in particular under-the-hood JIT compilation, making it both fully compiled and fully interactive. In terms of the more fundamental differences between "low-level" and "high-level", Julia chooses an unconventional approach: automatic memory management, but data types at the machine level.
As an illustration, consider integer handling. Julia's default integers are the same as C's: optimal machine-size signed integers with no overflow checks on arithmetic. The result of 10^50 is -5376172055173529600, for example. This is the best choice for performance, but it should be clear that it can easily create bugs. Traditional high-level languages use unlimited integers by default, eventually offering machine-size integers as a optimization option for experienced programmers. Julia does have a BigInt type, but using it requires a careful insertion of big(...) in many places. It's there if you absolutely need it, but you are expected to use machine-sized integers most of the time.
As a consequence, Julia is a power tool for experienced scientific programmers who are aware of the traps and the techniques to avoid falling into them. Julia is not a language suitable for beginners or occasional users of scientific programming, because such inexperienced scientists need more of a safety net than Julia provides. Neither is Julia a prototyping language for trying out new ideas, because when concentrating on the science you also need a safety net that protects you from the traps of machine-level abstractions. In Julia, you have to design your own safety net, and you also have to verify that it is strong enough for your needs.
Perhaps the biggest problem with Julia is that this is not obvious at first glance. Julia comes with all the nice interactive tools for rapid development and interactive data analysis, in particular the IJulia notebook which is basically the same as the now-famous IPython/Jupyter notebook. At a first glance, Julia looks like a traditional high-level language. A strong point of David's Julia tutorial is that it points out right from the start that Julia is different. Whenever a choice must be made between run-time efficiency and simplicity, clarity, or correctness, Julia always chooses efficiency. The least important consequence is surprising error messages that make sense only with a basic understanding of how the compiler works. The worst consequence is that inexperienced users are easily induced to write unsafe code. There are nice testing tools, in particular FactCheck which looks very nice, but scientists are notoriously unaware of the need of testing.
The worst design decision I see in Julia is the explicit platform dependence of the language: the default integer size is either 32 or 64 bits, depending on the underlying platform. This default size is used in particular for integer constants. As a consequence, a Julia program does in general not have a single well-defined result, but two distinct results. This means that programs must be tested on two different architectures, which is hard to do even for experienced programmers. Given the ongoing very visible debate about the (non-)reproducibility of computational research, I cannot understand how anyone can make such a decision today. Of course I do understand the performance advantage that results from this choice, but this clearly goes to far for my taste. If I ever use Julia for my research, I'll start each source code file with @assert WORD_SIZE==64 just to make sure that everyone knows what kind of machine I tested my code on.
As for the surprising but not dangerous features that can probably only be explained by convenience for the compiler, there is first of all the impossibility to redefine a data type without clearing the workspace first - and that means losing your whole session. It's a bit of a pain for interactive development, in particular in IJulia notebooks. Another oddity is the const declaration, which makes a variable to which you can assign new values as often as you like, as long as the type remains the same. It's more a typed variable declaration than the constant suggested by the name.
Finally, there is another point where I think the design for speed has gone too far. The choice of machine-size integers turns into something completely useless (in my opinion) when it comes to rational arithmetic. Julia lets you create fractions by writing 3//2 etc., but the result is a fraction whose nominator and denominator are machine-size integers. Rational arithmetic has the well-known performance and memory problem of denominators growing with each additional operation. With machine-size integers, rational arithmetic rapidly crashes or returns wrong results. Given that the primary application of rationals is unlimited precision arithmetic, I don't see a practical use for anything but Rational{BigInt}.
In the end, Julia leaves me with a feeling of a lost opportunity. My ideal software development environment for computational science would support the whole life cycle of computational methods, starting from prototyping and ending with platform-specific optimizations. As code is progressively optimized based on profiling information, each version would be used as a reference to test the next optimization level. In terms of fundamental language design, Julia seems to have everything required for such an approach. However, the default choice of fast-and-unsafe operations almost forces programmers into premature optimization. Like in the traditional high-/low-level language world, computational science will require two distinct languages, a safe and a fast one.
In a recent blog post, Titus Brown asks if software is a primary product of science, and basically says "no" (but do read the post for the details). A blog-post length reply by Daniel Katz comes to the opposite conclusion (again, please read the post before continuing here). I left a short comment on Titus' blog but also felt compelled to expand this into a blog post of its own - so here it is.
Titus introduces a useful criterion for what "primary product of science" is: could you get a Nobel prize for it? As Dan comments, Nobel prizes in science are awarded for discoveries and inventions. There we no computers when Alfred Nobel set up his foundation, so we have to extrapolate this definition a bit to today's situation. Is software like a discovery? Clearly not. Like an invention? Perhaps, but it doesn't fit very well. Dan makes a comparison with scientific writing, i.e. papers, textbooks, etc. Scientific writing is the traditional way to communicate discoveries and inventions. But what scientists get Nobel prizes for is not the papers, but the work described therein. Papers are not primary products of science either, they are just a means of communication. There is a fairly good analogy between papers and their contents on one hand, and software and algorithms on the other hand. And algorithms are very well comparable to discoveries and inventions. Moreover, many of today's scientific models are in fact expressed as algorithms. My conclusion is that algorithms clearly count as a primary product of science, but software doesn't. Software is a means of communication, just like papers or textbooks.
The analogy isn't perfect, however. The big difference between a paper and a piece of software is that you can feed the latter into a computer to make it do something. Software is thus a scientific tool a well as a means of communication. In fact, today's computational science gives more importance to the tool aspect than to the communication aspect. The main questions asked about scientific software are "What does it do?" and "How efficient is it?" When considering software as a means of communication, we would ask questions such as "Is it well-written, clear, elegant?", "How general is the formulation?", or "Can I use it as the basis for developing new science?". These questions are beginning to be heard, in the context of the scientific software crisis and the need for reproducible research. But they are still second thoughts. We actually accept as normal that the scientific contents of software, i.e. the models implemented by it, are understandable only to software specialists, meaning that for the majority of users, the software is just a black box. Could you imagine this for a paper? "This paper is very obscure, but the people who wrote it are very smart, so let's trust them and base our research on their conclusions." Did you ever hear such a claim? Not me.
Scientists haven't yet fully grasped the particular status of software as both an information carrier and a tool. That may be one of the few characteristics they share with lawyers. The latter make a difference between "data" (including written text), which is covered by copyright, and "software", which is covered by both copyright and licenses, and in some countries also by patents. Superficially, this makes sense, as it reflects the dual nature of software. It suffers, however, from two problems. First of all, the distinction exists only in the intention of the author, which is hard to pin down. Software is just data that can be interpreted as instructions for a computer. One could conceivably write some interpreter that turns previously generated data into software by executing it. Second, and that's a problem for science, the licensing aspect of software is much more restrictive than the copyright aspect. If you describe an algorithm informally in a paper, you have to deal only with copyright. If you communicate it in executable form, you have to worry about licensing and patents as well, even if your main intention is more precise communication.
I have written a detailed article about the problems resulting from the badly understood dual nature of scientific software, which I won't repeat here. I have also proposed a solution, the development of formal languages for expressing complex scientific models, and I am experimenting with a concrete approach to get there. I mention this here mainly to motivate my conclusion:
Q: Is software a primary product of science?
A: No. But neither is a paper or a textbook.
Q: Is software a means of communication for primary products of science?
A: Yes, but it's a bad one. We need something better.
While reading the final report of the reproducibility workshop at XSEDE14, I noticed a statement that I encounter frequently in discussions about reproducible research:
"One general consensus was that bitwise reproducibility is often an unrealistic expectation"
In the interest of clarity, let me start by pointing out that within the systematic terminology that I am trying to adopt (see this post for an explanation), I will write "bitwise replicability" from now on, as the problem falls into the technical domain (getting the same result from running the same program on the same data) rather than into the scientific one (verifying a result with similar but not identical methods and tools).
The particularity of bitwise replicability is that is almost always brushed aside as "unrealistic", which prevents any discussion about its possible importance in computational science. The main point of this post is to explain why I consider bitwise replicability important, but first of all I need to get the label "unrealistic" out of the way.
"Unrealistic" means more or less "possible in principle but impossible given various real-life contraints", and therefore the term should always be qualified by listing the constraints that make something impossible. In the context of bitwise replicability, which always refers to floating-point computations, the main constraint is that floating-point arithmetic is incompletely specified in most of today's programming languages, and that whatever specification there is is incompletely implemented in many of today's compilers. This is a valid reason for proclaiming bitwise replicability unrealistic for a short-term research project, but it is not an insurmountable barrier on a longer time scale. All we need are tighter specifications and implementations that respect them. That's a lot of work, but not a technical challenge. We know how to do it, but we are not (yet) willing to invest the effort to make it happen.
The main reason why I consider bitwise replicability important is software testing. No matter what precise approach is used for testing, it always involves comparing results of computations, either to a known good result, or to the result of another, presumably more reliable, computation. For any application of computing other than number crunching, comparing results means testing for equality, at the bit level. The results are equal or they aren't. If they aren't, there's a reason. You have to figure out what that reason is, and fix the problem.
If you accept the idea that floating-point operations are only approximate, the notion of a computation having one and only one result disappears, and testing becomes impossible. If two computations lead to similar but slightly different results, how do you decide if this is due to a bug or to some "inevitable" fuzziness of floating-point arithmetic? The answer is that you can't. If you accept that bitwise replicability is not possible, you also accept that rigorous software testing is not possible. For some illustrations of this problem, and some interesting discussion around them, see this post on the Software Carpentry blog.
The most common counterargument is that numerical methods are only approximate, that floating-point arithmetic is approximate as well, and that the main source of error comes from these two sources. That may or may not be true in any specific situation, as it really depends on what you are computing. But my point is that this statement can only be true if you assume that the implementation of your method contains no mistakes. The amount of error introduced by a bug in the code is completely unbounded. And even if it's small for some particular test run, it can be very large elsewhere. There is not much point in worrying about the error in an approximate numerical method unless you have some confidence in your code actually implementing this method correctly.
In fact, the common counterargument discussed above conflates several sources of error, which can and should be discussed and analyzed separately. A typical numerical computation is the result of several steps, starting from a mathematical model that takes the form of algebraic or differential equations:
Construct a computable approximation1 to the original equations, using techniques such as discretization of continuous quantities.
Replace real-numbers by floating-point numbers.
Implement the floating-point version in software.
The errors introduced in the first step are the subject of numerical analysis, a well-established domain of applied mathematics. They are well understood for most commonly employed numerical methods. The errors introduced in the second step are rarely discussed explicitly, outside of a small circle of researchers interested in the peculiarities of floating-point arithmetic. The third step should not introduce any errors, and that should be verified by testing. But uncoupling steps 2 and 3 is possible only if our software tools guarantee bitwise replicability.
So why don't today's tools permit this? The reason is a mixture of widespread ignorance about floating-point arithmetic and the desire to get maximum performance. Both come into play in step 2, which is approximating discrete equations for real numbers by discrete equations for floating-point numbers. Most scientific programmers are unaware that this is an approximation that they should understand and control. They just type their real-number equation into a program and expect the computer to handle it somehow. Compiler writers and language specification authors take advantage of this ignorance and declare this step their business, profiting from the many optimization possibilities it offers.
The optimization opportunities come from the fact that a typical real-number equation has a large number of a priori equally plausible floating-point number approximations. Many of the identities for real numbers do not apply to floating-point numbers, for example associativity of addition and multiplication. Where the real-number equation says a+b+c, there are three floating-point approximations: (a+b)+c, a+(b+c), and (a+c)+b. For more complex equations, the number of variants quickly becomes important. The results of these variants are not the same, but which one to choose? The choice should be made after a careful analysis of the relative precision and performance of each variant. There should be tool support to help with this. But what happens in practice, most of the time, is that the choice is made by the compiler, which goes exclusively for performance. Since every compiler optimizes differently, the same program source code yields different results on different platforms. And that's why we don't have bitwise replicability.
To prevent any misunderstanding: I am not saying that production-level compiled code needs to ensure bitwise reproducibility across machines. It's OK to have compiler optimization options that introduce platform-specific approximations. But it should be possible to reproduce one unique result identically on all platforms. This result is then the reference against which additional "lossy" optimizations can be tested.
Footnotes:
1 I am using the term "computable approximation" somewhat vaguely here. While the original continuous-variable equations are almost always non-computable, and the numerical approximations are mostly computable, there are exceptions on both sides. The main focus of numerical analysis is not computability in the strict sense of computability theory, but "practical" computability that has the subsequent transformation to floating-point operations in mind.
The importance of reproducibility in computational science is being more and more recognized, which I think is a good sign. However, I also notice a lot of confusion about what reproducibility means exactly, and also confusion about the difference (if any) between reproducibility and replicability. I don't see a consensus yet about the exact meaning of these terms, but I would like to give my own definitions and justify them by putting them into the general context of computational science.
I'll start with the concept of reproducibility as it was used in science long before computers even existed. It refers to the reproducibility of the conclusions of a scientific study. These conclusions can take very different forms depending on the question that was being explored. It can be a simple "yes" or "no", e.g. in answering questions such as "Is the gravitational force acting in this stone the same everywhere on the Earth's surface?" or "Does ligand A bind more strongly to protein X than ligand B?" It can also be a number, as in "What is the lattice energy of NaCl?", or a mathematical function, as in "How does a spring's restoring force vary with elongation?" Any such result should come with an estimation of its precision, such as an error bar on numbers, or a reliability estimate for a yes/no answer. Reproducing a scientific conclusion means finding a "close enough" answer by performing "similar" experiments and analyses. As the terms "close enough" and "similar" show, reproducibility involves human judgement, which may well evolve over time. Reproducibility is thus not an absolute feature of a specific result, but the evaluation of a result in the context of the current state of knowledge and technology in a scientific domain. Every attempt to reproduce a given result independently (different people, tools, methods, …) augments scientific knowledge: If the reproduction leads to a "close enough" results, it provides information about the precision with which the results can be obtained, and if if doesn't, it points to some previously unrecognized crucial difference between the two experiments, which can then be explored.
Replication refers to something much more specific: repeating the exact steps in an experiment using the same (or equivalent) equipment, and comparing the outcomes. Replication is part of testing an experimental setup, or a form of quality assurance. If I measure the same quantity ten times using the same equipment and experimental samples, and get ten slightly different values, then I can use these numbers to estimate the precision of my equipment. If that precision is not sufficient for the purposes of my planned scientific study, then the equipment is not suitable.
It is useful to describe the process of doing research by a two-layer model. The fundamental layer is the technology layer: equipment and procedures that are well understood and whose precision is known from many replication attempts. On top of this, there is the research layer: the well-understood equipment is used in order to obtain new scientific information and draw conclusions from them. Any scientific project aims at improving one or the other layer, but not both at the same time. When you want to get new scientific knowledge, you use trusted equipment and procedures. When you want to improve the equipment or the procedures, you do so by doing test measurements on well-known systems. Reproducibility is a concept of the research layer, replicability belongs to the technology layer.
All this carries over identically to computational science, in principle. There is the technology layer, consisting of computers and the software that runs on them, and the research layer, which uses this technology to explore theoretical models or to interpret experimental data. Replicability belongs to the technology level. It increases trust in a computation and thus its components (hardware, software, overall workflow, provenance tracking, …). If a computation cannot be replicated, then this points to some kind of problem:
different input data that was not recorded in the workflow (interactive user input, a random number stream initialized from the current time, …)
a bug in the software (uninitialized variables, compiler bugs, …)
a fault in the hardware (an unreliable memory chip, a design flaw in the processor, …)
an ambiguous specification of the result of the computation
Ideally, the non-replicability should be eliminated, but at the very least its cause should be understood. This turns out to be very difficult in practice, in today's computing environments, essentially because case 4 is frequent and hard to avoid (today's popular programming languages are ambiguous), and because case 4 makes it impossible to identify cases 2 and 3 with certainty. I see this as a symptom of the immaturity of today's computing environments, which the computational science community should aim to improve on. The technology for removing case 4 exists. The keyword is "formal methods", and there are first attempts to apply them to scientific computing, but this remains an exotic approach for now.
As in experimental science, reproducibility belongs to the research layer and cannot be guaranteed or verified by any technology. In fact, the "reproducible research" movement is really about replicability - which is perhaps one reason for the above-mentioned confusion.
There is at the moment significant disagreement about the importance of replicability. At one end of the spectrum, there is for example Ian Gent's recomputation manifesto, which stresses the importance of replicability (which in the context of computational science he calls recomputability) because building on past work is possible only if it can be replicated as a first step. At the other end, Chris Drummond argues that replicability is "not worth having" because it doesn't contribute much to the real goal, which is reprodcucibility. It is worth reading both of these papers, because they both do a very good job at explaining their arguments. There is actually no contradiction between the two lines of arguments, the different conclusions are due to different criteria being applied: Chris Drummond sees replicability as valuable only if it improves reproducibility (which indeed it doesn't), whereas Ian Gent sees value in it for a completely different reason: it makes future research more efficient. Neither one mentions the main point in favor of replicability that I have made above: that replicability is a form of quality assurance and thus increases trust in published results.
It is probably a coincidence that both of the papers cited above use the term "computational experiment", which I think should best be avoided in this context. In the natural sciences, the term "experiment" traditionally refers to constructing a setup to observe nature, which makes experiments the ultimate source of truth in science. Computations do not have this status at all: they are applications of theoretical models, which are always imperfect. In fact, there is an interesting duality between the two: experiments are imperfect observations of the ultimate truth, whereas computations are, in the absence of buggy or ambiguous software, perfect observations of the consequences of imperfect models. Using the same term for these two concepts is a source of confusion, as I have pointed out earlier.
This fundamental difference between experiments and computations also means that replicability has a different status in experimental and computational science. When doing imperfect observations of nature, evaluating replicability is one aspect of evaluating the imperfection of the observation. Perfect observation is impossible, both due to technological limitations and for fundamental reasons (any observation modifies what is being observed). On the other hand, when computing the consequences of imperfect models, replicability does not measure the imperfections of the model, but the imperfections of the computation, which can theoretically be eliminated.
The main source of imperfections in computations is the complexity of computer software (considering the whole software stack, from the operating system to the scientific software). At this time, it is not clear if we will ever succeed in taming this complexity. Our current digital computers are chaotic systems, in which even the tiniest change (flipping a bit in memory, or replacing a single character in a program source code file) can change the result of a computation beyond any bounds. Chaotic behavior is clearly an undesirable feature in any scientific equipment (I can't think of any experimental apparatus suffering from it), but for computation we currently have no other choice. This makes quality assurance techniques, including replicability but also more standard software engineering practices such as unit testing, all the more important if we want computational results to be trustworthy.
Over the last few months I have been exploring the Racket language for its potential as a language for computational science, and it's time to summarize my first impressions.
Why Racket?
There are essentially two reasons for learning a programing language: (1) getting acquainted with a new tool that promises to get some job done better than with other tools, and (2) learning about other approaches to computing and programming. My interest in Racket was driven by a combination of these two aspects. My background is in computational science (phsyics, chemistry, and structural biology), so I use computation extensively in my work. Like most computational scientists of my generation, I started working in Fortran, but quickly found this unsatisfactory. Looking for a better way to do computational science, I discovered Python in 1994 and joined the Matrix-SIG that developed what is now known as NumPy. Since then, Python has become my main programming language, and the ecosystem for scientific computing in Python has flourished to a degree unimaginable twenty years ago. For doing computational science, Python is one of the top choices today.
However, we shouldn't forget that we are still living in the stone age of computational science. Fortran was the Paleolithic, Python is the Neolithic, but we have to move on. I am convinced that computing will become as much an integral part of doing science as mathematics, but we are not there yet. One important aspect has not evolved since the beginnings of scientific computing in the 1950s: the work of a computational scientist is dominated by the technicalities of computing, rather than by the scientific concerns. We write, debug, optimize, and extend software, port it to new machines and operating systems, install messy software stacks, convert file formats, etc. These technical aspects, which are mostly unrelated to doing science, take so much of our time and attention that we think less and less about why we do a specific computation, how it fits into more general theoretical frameworks, how we can verify its soundness, and how we can improve the scientific models that underly our computations. Compare this to how theoreticians in a field like physics or chemistry use mathematics: they have acquired most of their knowledge and expertise in mathematics during their studies, and spend much more time applying mathematics to do science than worrying about the intrinsic problems of mathematics. Computing should one day have the same role. For a more detailed description of what I am aiming at, see my recent article.
This lengthy foreword was necessary to explain what I am looking for in Racket: not so much another language for doing today's computational science (Python is a better choice for that, if only for its well-developed ecosystem), but as an evironment for developing tomorrow's computational science. The Racket Web site opens with the title "A programmable programming language", and that is exactly the aspect of Racket that I am most interested in.
There are two more features of Racket that I found particularly attractive. First, it is one of the few languages that have good support for immutable data structures without being extremist about it. Mutable state is the most important cause of bugs in my experience (see my article on "Managing State" for details), and I fully agree with Clojure's Rich Hickey who says that "immutability is the right default". Racket has all the basic data structures in a mutable and an immutable variant, which provides a nice environment to try "going immutable" in practice. Second, there is a statically typed dialect called Typed Racket which promises a straightforward transition from fast prototyping in plain Racket to type-safe and more efficient production code in Typed Racket. I haven't looked at this yet, so I won't say any more about it.
Racket characteristics
For readers unfamiliar with Racket, I'll give a quick overview of the language. It's part of the Lisp family, more precisely a derivative of Scheme. In fact, Racket was formerly known as "PLT Scheme", but its authors decided that it had diverged sufficiently from Scheme to give it a different name. People familiar with Scheme will still recognize much of the language, but some changes are quite profound, such as the fact that lists are immutable. There are also many extensions not found in standard Scheme implementations.
The hallmark of the Lisp family is that programs are defined in terms of data structures rather than in terms of a text-based syntax. The most visible consequence is a rather peculiar visual aspect, which is dominated by parentheses. The more profound implication, and in fact the motivation for this uncommon choice, is the equivalence of code and data. Program execution in Lisp is nothing but interpretation of a data structure. It is possible, and common practice, to construct data structures programmatically and then evaluate them. The most frequent use of this characteristic is writing macros (which can be seen as code preprocessors) to effectively extend the language with new features. In that sense, all members of the Lisp family are "programmable programming languages".
However, Racket takes this approach to another level. Whereas traditional Lisp macros are small code preprocessors, Racket's macro system feels more like a programming API for the compiler. In fact, much of Racket is implemented in terms of Racket macros. Racket also provides a way to define a complete new language in terms of existing bits and pieces (see the paper "Languages as libraries" for an in-depth discussion of this philosophy). Racket can be seen as a construction kit for languages that are by design interoperable, making it feasible to define highly specific languages for some application domain and yet use it in combination with a general-purpose language.
Another particularity of Racket is its origin: it is developed by a network of academic research groups, who use it as tool for their own research (much of which is related to programming languages), and as a medium for teaching. However, contrary to most programming languages developed in the academic world, Racket is developed for use in the "real world" as well. There is documentation, learning aids, development tools, and the members of the core development team are always ready to answer questions on the Racket user mailing list. This mixed academic-application strategy is of interest for both sides: researchers get feedback on the utility of their ideas and developments, and application programmers get quick access to new technology. I am aware of only three other languages developed in a similar context: OCaml, Haskell, and Scala.
Learning and using Racket
A first look at the Racket Guide (an extended tutorial) and the Racket Reference shows that Racket is not a small language: there is a bewildering variety of data types, control structures, abstraction techniques, program structuration methods, and so on. Racket is a very comprehensive language that allows both fine-tuning and large-scale composition. It definitely doesn't fit into the popular "low-level" vs. "high-level" dichotomy. For the experienced programmer, this is good news: whatever technique you know to be good for the task at hand is probably supported by Racket. For students of software development, it's probably easy to get lost. Racket comes with several subsets developed for pedagogical purposes, which are used in courses and textbooks, but I didn't look at those. What I describe here is the "standard" Racket language.
Racket comes with its own development environment called "DrRacket". It looks quite poweful, but I won't say more about it because I haven't used it much. I use too many languages to be interested in any language-specific environment. Instead, I use Emacs for everything, with Geiser for Racket development.
The documentation is complete, precise, and well presented, including a pleasant visual layout. But it is not always an easy read. Be prepared to read through some background material before understanding all the details in the reference documentation of some function you are interested in. It can be frustrating sometimes, but I have never been disappointed: you do find everything you need to know if you just keep on following links.
My personal project for learning Racket is an implementation of the MOSAIC data model for molecular simulations. While my implementation is not yet complete (it supports only two kinds of data items, universes and configurations), it has data structure definitions, I/O to and from XML, data validation code, and contains a test suite for everything. It uses some advanced Racket features such as generators and interfaces, not so much out of necessity but because I wanted to play with them.
Overall I had few surprises during my first Racket project. As I already said, finding what you need in the documentation takes a lot of time initially, mostly because there is so much to look at. But once you find the construct you are looking for, it does what you expect and often more. I remember only one ongoing source of frustration: the multitude of specialized data structures, which force you to make choices you often don't really care about, and to insert conversion functions when function A returns a data structure that isn't exactly the one that function B expects to get. As an illustration, consider the Racket equivalent of Python dictionaries, hash tables. They come in a mutable and an immutable variant, each of which can use one of three different equality tests. It's certainly nice to have that flexibility when you need it, but when you don't, you don't want to have to read about all those details either.
As for Racket's warts, I ran into two of them. First, the worst supported data structure in Racket must be the immutable vector, which is so frustrating to work with (every operation on an immutable vector returns a mutable vector, which has to be manually converted back to an immutable vector) that I ended up switching to lists instead, which are immutable by default. Second, the distinction (and obligatory conversion) between lists, streams, generators and a somewhat unclear sequence abstraction makes you long for the simplicity of a single sequence interface as found in Python or Clojure. In Racket, you can decompose a list into head and tail using first and rest. The same operations on a stream are stream-first and stream-rest. The sequence abstraction, which covers both lists and streams and more, has sequence-tail for the tail, but to the best of my knowledge nothing for getting the first element, other than the somewhat heavy (for/first ([element sequence]) element).
The macro requirements of my first project were modest, not exceeding what any competent Lisp programmer would easily do using defmacro (which, BTW, exists in Racket for compatibility even though its use is discouraged). Nevertheless, in the spirit of my exploration, I tried all three levels of Racket's hygienic macro definitions: syntax-rule, syntax-case, and syntax-parse, in order of increasing power and complexity. The first, syntax-rule is straightforward but limited. The last one, syntax-parse, is the one you want for implementing industrial-strength compiler extensions. I don't quite see the need for the middle one, syntax-case, so I suppose it's there for historical reasons, being older than syntax-parse. Macros are the one aspect of Racket for which I recommend starting with something else than the Racket documentation: Greg Hendershott's Fear of Macros is a much more accessible introduction.
Scientific computing
As I said in the beginning of this post, my goal in exploring Racket was not to use it for my day-to-day work in computational science, but nevertheless I had a look at the support for scientific computing that Racket offers. In summary, there isn't much, but what there is looks very good.
The basic Racket language has good support for numerical computation, much of which is inherited from Scheme. There are integers of arbitrary size, rational numbers, and floating-point numbers (single and double precision), all with the usual operations. There are also complex numbers whose real/imaginary parts can be exact (integer or rational) or inexact (floats). Unlimited-precision floats are provided by an interface to MPFR in the Racket math library.
The math library (which is part of every standard Racket installation) offers many more goodies: multidimensional arrays, linear algebra, Fourier transforms, special functions, probability distributions, statistics, etc. The plot library, also in the standard Racket installation, adds one of the nicest collections of plotting and visualization routines that I have seen in any language. If you use DrRacket, you can even rotate 3D scenes interactively, a feature that I found quite useful when I used (abused?) plots for molecular visualization.
Outside of the Racket distribution, the only library I could find for scientific applications is Doug Williams' "science collection", which predates the Racket math library. It looks quite good as well, but I didn't find an occasion yet for using it.
Could I do my current day-to-day computations with Racket? A better way to put it is, how much support code would I have to write that is readily available for more mature scientific languages such as Python? What I miss most is access to my data in HDF5 and netCDF formats. And the domain-specific code for molecular simulation, i.e. the equivalent of my own Molecular Modeling Toolkit. Porting the latter to Racket would be doable (I wrote it myself, so I am familiar with all the algorithms and its pitfalls), and would in fact be an opportunity to improve many details. But interfacing HDF5 or netCDF sounds like a lot of work with no intrinsic interest, at least to me.
The community
Racket has an apparently small but active, competent, and friendly community. I say "apparently" because all I have to base my judgement on is the Racket user mailing list. Given Racket's academic and teaching background, it is quite possible that there are lots of students using Racket who find sufficient support locally that they never manifest themselves on the mailing list. Asking a question on the mailing list almost certainly leads to a competent answer, sometimes from one of the core developers, many of whom are very present. There are clearly many Racket beginners (and also programming newbies) on the list, but compared to other programming language users' lists, there are very few naive questions and comments. It seems like people who get into Racket are serious about programming and are aware that problems they encounter are most probably due to their lack of experience rathen than caused by bugs or bad design in Racket.
I also noticed that the Racket community is mostly localized in North America, judging from the peak posting times on the mailing list. This looks strange in today's Internet-dominated world, but perhaps real-life ties still matter more than we think.
Even though the Racket community looks small compared to other languages I have used, it is big and healthy enough to ensure its existence for many years to come. Racket is not the kind of experimental language that is likely to disappear when its inventor moves on to the next project.
Conclusion
Overall I am quite happy with Racket as a development language, though I have to add that I haven't used it for anything mission-critical yet. I plan to continue improving and completing my Racket implementation of Mosaic, and move it to Typed Racket as much as possible. But I am not ready to abandon Python as my workhorse for computational science, there are simply too many good libraries in the scientific Python ecosystem that are important for working efficiently.
Why do people write computer programs? The answer seems obvious: in order to produce useful tools that help them (or their clients) do whatever they want to do. That answer is clearly an oversimplification. Some people write programs just for the fun of it, for example. But when we replace "people" by "scientists", and limit ourselves to the scientists' professional activities, we get a statement that rings true: Scientists write programs because these programs do useful work for them. Lengthy computations, for example, or visualization of complex data.
This perspective of "software as a tool for doing research" is so pervasive in computational science that it is hardly ever expressed. Many scientists even see software, or perhaps the combination of computer hardware plus software as just another piece of lab equipment. A nice illustration is this TEDx lecture by Klaus Schulten about his "computational microscope", which is in fact Molecular Dynamics simulation software for studying biological macromolecules such as proteins or DNA.
To see the fallacy behind equating computer programs with lab equipment, let's take a step back and look at the basic principles of science. The ultimate goal of science is to develop an understanding of the universe that we inhabit. The specificity of science (compared to other approaches such as philosophy or religion) is that it constructs precise models for natural phenomena that it validates and improves by repeated confrontation with observations made on the real thing:
An experiment is just an optimization: it's a setup designed for making a very specific kind of observation that might be difficult or impossible to make by just looking at the world around us. The process of doing science is an eternal cycle: the model is used to make predictions of yet-to-make observations, whereas the real observations are compared to these predictions in order to validate the model and, in case of a significant discrepancies, to correct it.
In this cycle of prediction and observation, the role of a traditional microscope is to help make observations of what happens in nature. In contrast, the role of Schulten's computational microscope is to make predictions from a theoretical model. Once you think about this for a while, it seems obvious. To make observations on a protein, you need to have that protein. A real one, made of real atoms. There is no protein anywhere in a computer, so a computer cannot do observations on proteins, no matter which software is being run on it. What you look at with the computational microscope is not a protein, but a model of a protein. If you actually watch Klaus Schulten's video to the end, you will see that this distinction is made at some point, although not as clearly as I think it should be.
So it seems that the term "a tool for exploring a theoretical model" is a good description of a simulation program. And in fact that's what early simulation programs were. The direct ancestors of Schulten's computational microscope are the first Molecular Dynamics simulation programs made for atomic liquids. A classic reference is Rahman's 1964 paper on the simulation of liquid argon. The papers of that time specify the model in terms of a few mathematical equations plus a some numerical parameters. Molecular Dynamics is basically Newton's equations of motion, discretized for numerical integration, plus a simple model for the interactions between the atoms, known as the Lennard-Jones potential. A simulation program of the time was a rather straightforward translation of the equations into FORTRAN, plus some bookkeeping and I/O code. It was indeed a tool for exploring a theoretical model.
Since then, computer simulation has been applied to ever bigger and ever more complex systems. The examples shown by Klaus Schulten in his video represent the state of the art: assemblies of biological macromolecules, consisting of millions of atoms. The theoretical model for these systems is still a discretized version of Newton's equations plus a model for the interactions. But this model for the interactions has become extremely complex. So complex in fact that nobody bothers to write it down any more. It's not even clear how you would write it down, since standard mathematical notation is no longer adequate for the task. A full specification requires some algorithms and a database of chemical information. Specific aspects of model construction have been discussed at length in the scientific literature (for example how best to describe electrostatic interactions), but a complete and precise specification of the model used in a simulation-based study is never provided.
The evolution from simple simulations (liquid argon) to complex ones (assemblies of macromolecules) looks superficially like a quantitative change, but there is in fact a qualitative difference: for today's complex simulations, the computer program is the model. Questions such as "Does program X correctly implement model A?", a question that made perfect sense in the 1960s, have become meaningless. Instead, we can only ask "Does program X implement the same model as program Y?", but that question is impossible to answer in practice. The reason is that the programs are even more complex than the models, because they also deal with purely practical issues such as optimization, parallelization, I/O, etc. This phenomenon is not limited to Molecular Dynamics simulations. The transition from mathematical models to computational models, which can only be expressed in the form of computer programs, is happening in many branches of science. However, scientists are slow to recognize what is happening, and I think that is one reason for the frequent misidentification of software as experimental equipment. Once a theoretical model is complex and drowned in even more complex software, it acquires many of the characteristics of experiments. Like a sample in an experiment, it cannot be known exactly, it can only be studied by observing its behavior. Moreover, these observations are associated with systematic and statistical errors resulting from numerical issues that frequently even the program authors don't fully understand.
From my point of view (I am a theoretical physicist), this situation is not acceptable. Models play a central role in science, in particular in theoretical science. Anyone claiming to be theoretician should be able to state precisely which models he/she is using. Differences between models, and approximations to them, must be discussed in scientific studies. A prerequisite is that the models can be written down in a human-readable form. Computational models are here to stay, meaning that computer programs as models will become part of the daily bread of theoreticians. What we will have to develop is notations and techniques that permit a separation of the model aspect of a program from all the other aspects, such as optimization, parallelization, and I/O handling. I have presented some ideas for reaching this goal in this article (click here for a free copy of the issue containing it, it's on page 77), but a lot of details remain to be worked out.
The idea of programs as a notation for models is not new. It has been discussed in the context of education, for example in this paper by Gerald Sussman and Jack Wisdom, as well as in their book that presents classical mechanics in a form directly executable on a computer. The constraint of executability imposed by computer programs forces scientists to remove any ambiguities from their models. The idea is that if you can run it on your computer, it's completely specified. Sussman and Wisdom actually designed a specialized programming language for this purpose. They say it's Scheme, which is technically correct, but Scheme is a member of the Lisp family of extensible programming languages, and the extensions written by Sussman and Wisdom are highly non-trivial, to the point of including a special-purpose computer algebra system.
For the specific example that I have used above, Molecular Dynamics simulations of proteins, the model is based on classical mechanics and it should thus be possible to use the language of Sussman and Wisdom to write down a complete specification. Deriving an efficient simulation program from such a model should also be possible, but requires significant research and devlopment effort.
However, any progress in this direction can happen only when the computational science community takes a step back from its everyday occupations (producing ever more efficient tools for running ever bigger simulations on ever bigger computers) and starts thinking about the place that it occupies in the pursuit of scientific research.
This post was motivated by Ian Gent's recomputation manifesto and his blog post about it. While I agree with pretty much everything said there, there is one point that I strongly disagree with, and here I'd like to explain the reasons in some detail. The point in question is "The only way to ensure recomputability is to provide virtual machines". To be fair, the manifesto specifies that it's the only way "at least for now", so perhaps our disagreement is not as pronounced as it may seem.
I'll start with a quote from the manifesto that shows that we have similar ideas of the time scales over which computational research should be reproducible: "It may be true that code you make available today can be built with only minor pain by many people on current computers. That is unlikely to be true in 5 years, and hardly credible in 20."
So the question is: how can we best ensure that the software used in our computational studies can still be run, with reasonable effort, 20 years from now. To answer that question, we have to look at the possible platforms for computational research.
By a "platform", I mean the combination of hardware and software that is required to use a given piece of digital information. For example, Flash video requires a Flash player and a computer plus operating system that the Flash player can run on. That's what defines the "Flash platform". Likewise, today's "Web platform" (a description that requires a date stamp to be precise, because Web standards evolve so quickly) consists of HTML5, JavaScript, and a couple of related standards. If you want to watch a Flash video in 20 years, you will need a working Flash platform, and if you want to use an archived copy of a 2013 Web site, you need the 2013 Web platform.
If you plan to distribute some piece of digital information with the hope that it will make sense 20 years from now, you must either have confidence in the longevity of the platform, or be willing and able to ensure its long-term maintenance yourself. For the Flash platform, that means confidence in Adobe and its willingness to keep Flash alive (I wouldn't bet on that). For the 2013 Web platform, you may hope that its sheer popularity will motivate someone to keep it alive, but I wouldn't bet on it either. The Web platform is too complex and too ill-defined to be kept alive reliably when no one uses it in daily life any more.
Back to computational science. 20 years ago, most scientific software was written in Fortran 77, often with extensions specific to a machine or compiler. Much software from that era relied on libraries as well, but they were usually written in the same language, so as long as their source code remains available, the platform for all that is a Fortran compiler compatible with the one from back then. For standard Fortran 77, that's not much of a problem, whereas most of the vendor-specific extensions have disappeared since. Much of that 20-year-old software can in fact still be used today. However, reproducing a computational study based on that software is a very different problem: it also requires all the input data and an executable description of the computational protocol. Even in the rare case that all that information is available, it is likely to depend on lots of other software pieces that may not be easy to get hold of any more. The total computational platform for a given research project is in fact as ill-defined as the 2013 Web platform.
Today's situation is worse, because we use more diverse software written in more different languages, and also use more interactive software whose use is notoriously non-reproducible. The only aspect where we have gained in standardization is the underlying hardware and OS layer: pretty much all computational science is done today on x86 processors running Linux. Hence the idea of conserving the full operating environment in the form of a virtual machine. Just fire up VirtualBox (or one of the other virtual machine managers) and run an exact copy of the original study's work environment.
But what is the platform required to run today's virtual machines? It's VirtualBox, or one of its peers. Note however that it's not "any of today's virtual machine managers" because compatibility between their virtual machine formats is not perfect. It may work, or it may not. For simplicity I will use VirtualBox in the following, but you can substitute another name and the basic arguments still hold.
VirtualBox is a highly non-trivial piece of software, and it has very stringent hardware requirements. Those hardware requirements are met by the vast majority of today's computing equipment used in computational science, but the x86 platform is losing market share rapidly on the wider computing device market. VirtualBox doesn't run on an iPad, for example, and probably it never will. Is VirtualBox likely to be around in 20 years? I won't dare a prediction. If x86 survives for another 20 years AND if Oracle sees a continuing interest in this product, then it will. I won't bet on it though.
What we really need for long-term recomputability is a simple platform. A platform that is simple enough that the scientific community alone can afford to keep it alive for its own needs, even if no one else in the world cares about it.
Unfortunately there is no suitable platform today, to the best of my knowledge. Which is why virtual machines are perhaps the best option right now, for lack of a satisfactory one. But if we care about recomputability, we should design and develop a good supporting platform, starting as soon as possible.
For a more detailed discussion of this issue, see this paper written by yours truly. It comes to the conclusion that the closest existing approximation to a good platform is the Java virtual machine. What we'd want ideally is something similar to the JVM, but designed and optimized for scientific applications. A basic JVM implementation is quite simple (the complex JIT stuff is not a requirement), a few orders of magnitude simpler than VirtualBox, and it has no specific hardware dependencies. It's even simpler than many of today's scientific software packages, so the scientific community can definitely afford to keep it alive, The tough part is... no, it's not designing or writing the required software, it's agreeing on a specification. Perhaps it will never happen. Perhaps virtual machines will remain the best choice for lack of a satisfactory one. Or perhaps we will end up compiling our software to asm.js and run in the browser, just because someone else will keep that platform alive for us, no matter how ill-adapted it is to our needs. But don't say you haven't been warned.
A while ago I was chatting with two users of my Molecular Modelling Toolkit (MMTK), a library for molecular simulations written in Python. One of them asked me what I would do differently if I were to write MMTK today. That's an interesting question, but not the kind of question I can answer in a sentence or two, so I promised to write a blog post about this. Here it is.
First, a bit of history. The first version of MMTK was released about 16 years ago. I don't have the exact data, but the first message on the MMTK mailing list, announcing MMTK release 1.0b2, is dated 29 May 1997. Back then Python 1.4 was the state of the art and Numerical Python was a young project that was just beginning to stabilize. MMTK was one of the first domain-specific scientific libraries written in Python, at a time when the scientific Python community was very small and its members were mostly considered cranks by their peers. MMTK was designed from the start as a Python library, with relatively small bits of C code for the time-critical stuff (mainly energy evaluation and MD integration), with NumPy arrays at the Python-C interface. This has since become one of the two main approaches to using Python in scientific computing, the other one being wrapper code around libraries written in C/C++ or Fortran.
So what would I do differently if I were to start writing MMTK today? Many things, for different reasons. Lets first get the obvious stuff out of the way: the Python ecosystem has evolved significantly since 1997, and of course I would use Python 3, and Cython instead of C for the time-critical parts. I would also adopt many of the conventions that the community has developed but which weren't around in 1997. I might even be tempted to use bleeding-edge tools like Numba, although with hesitation: Numba is not only a moving target at this time, but also requires dependencies (I am thinking mostly of LLVM) which are big and non-trivial to install. One lesson I have learned in 16 years of scientific Python is that dependencies can cause more trouble than they are worth. It's nice in theory to re-use existing tested code, but it also makes installation and deployment more cumbersome.
So far for changes in the Python ecosystem. What has changed as well, though at a slower pace, is the role of computation in science and in particular in molecular simulation. Back in 1997, there were a few molecular simulation ecosystems that operated almost in isolation. The big players were the CHARMM, AMBER, and GROMOS/GROMACS communities. Each of them had their own software, their own file formats, and their own force fields. Members of these communities would of course talk about science to each other, but not share any software or data. Developing new computational methods required a serious investment into one of these ecosystems. That was in fact my main motivation for developing MMTK: I figured that I would be more efficient (not to mention more satisfied) writing a new system from scratch using modern development tools than trying to get familiar with crufty Fortran code. But I adopted basically the same approach with MMTK: I created a new ecosystem without much regard to sharing code or data with the rest of the world. As an illustration, MMTK defines its own trajectory format which I still consider superior to what the rest of the world is doing, but which is undeniably hard to use without MMTK, given that the definition of a universe is stored as an executable Python expression. MMTK also encourages storing data as Python pickle files, which are even harder to deal with for other programs.
Today we are seeing a change in attitude in computational science that I am sure will soon reach the molecular simulation community as well. People are starting to realize that computational results have serious reliability problems. The most publicized case in the structural biology community was the retraction of a few important published protein structures following the discovery of a bug in the data processing software that lead to completely wrong final structures. This and similar events point to the urgent need for better validation of computational results. One aspect of validation is re-running the same computation with different tools. Another aspect is publishing both software and raw data, enabling other scientists to inspect them and check their validity. Technology for sharing scientific code and data exists today (have a look at Github, Bitbucket, and figshare, for example). But in molecular simulation, there are still important practical barriers to such validation attempts, in particular the use of program-specific and badly documented file formats. While MMTK's file formats are documented, they are still program-specific and thus incompatible with the requirements of the future.
The sentence that I would like to write now is "If I were to rewrite MMTK today, I would use the exchange data formats accepted by the molecular simulation community". But those formats don't exist yet, although there are a few initiatives to develop them. My own contribution to this effort is the Mosaic data model and data formats - if you are interested in this subject, please have a look at it and send me your feedback. Mosaic will of course find its way into future versions of MMTK.
Finally, there are things I would do differently because the experience with MMTK has shown that a few initial design decisions were not the best ones. Number one is the absence of stable atom numbers. In MMTK, each atom and molecule is represented by a unique Python object, and there are ways to refer uniquely to everything by using Python expressions. But there is no such thing as a unique order of atoms that would assign a number to each one. Atoms do have numbers by which the low-level C code refers to them, but these numbers can be different every time you run a Python script. My original design goal was to discourage the use of numbers to refer to atoms, because this is an important source of mistakes if the simulated system undergoes changes. But every other molecular simulation program out there uses numbers to refer to atoms, so people are used to them. For interoperability with other programs, atom numbers are fundamental. There are ways to handle such situations, of course, but it's a constant source of headaches.
The other design aspect that I would change if I were to rewrite MMTK today is the hierarchy of chemical objects. MMTK has Atoms, Groups, Molecules, and Complexes, plus specializations such as AminoAcidResidue (a special Group), PeptideChain (a special Molecule), and Protein (a special Complex). While all of these correspond to some chemical reality, the system is more complex than required for molecular simulation, leading in some situations to code that is bloated by irrelevant special cases. Today I'd go for just Atoms and Groups, with special features of specific kinds of groups indicated by attributes rather than specific classes.
Calculators are among the most popular applications for smartphones, and therefore it is not surprising that the Google Play Store has more than 1000 calculators for the Android platform. Having used HP's scientific calculators for more than 20 years, I picked RealCalc when I got my Android phone and set it to RPN mode. It works fine, I have no complaints about it. But I no longer use it because I found something much more powerful.
It's called "J", which isn't exactly a very descriptive name. And that's probably a good idea because describing it it not so easy. J is much more than a calculator, but it does the calculator job very well. It's actually a full programming language, but one that differs substantially from everything else that goes by that label. The best description for J I can come up with is "executable mathematical notation". You type an expression, and you get the result. That's in fact not very different from working interactively with Python or Matlab, except that the expressions are very different. You can write traditional programs in J, using loops, conditionals, etc., but you can a lot of work done without ever using these features.
The basic data structure in J is the array, which can have any number of dimensions. Array elements can be numbers, characters, or other arrays. Numbers (zero-dimensional arrays) and text strings (one-dimensional arrays of characters) are just special cases. In J jargon, which takes its inspiration from linguistics, data items are called "nouns". Standard mathematical operators (such as + or -) are called "verbs" and can have one or two arguments (one left, one right). An expression is called a "sentence". There are no precedence rules, the right argument of any verb being everything to its right. Given the large number of verbs in J, this initially unfamiliar rule makes a lot of sense. A simple example (also showing the use of arrays) is
2 * 3 + 10 20 30 26 46 66
Up to here, J expressions are not very different from Python or Matlab expressions. What J doesn't have is functions with the familiar f(x, y, z) syntax, accepting any number of arguments. There are only verbs, with one or two arguments. But what makes J really different from the well-known languages for scientific computing are the "parts of speech" that have no simple equivalent elsewhere: adverbs and conjunctions.
An adverb takes a verb argument and produces a derived verb from it. For example, the adverb ~ takes a two-argument verb (a dyad in J jargon) and turns it into a one-argument verb (a monad) that's equivalent to using the dyad with two equal arguments. With + standing for plain addition, +~ thus doubles its argument:
+~ 1 5 10 20 2 10 20 40
meaning it is the same as
1 5 10 20 + 1 5 10 20 2 10 20 40
A conjunction combines a verb with a noun or another verb to produce a derived verb. An example is ^:, the power conjunction, which applies a verb several times:
The parentheses are required to separate the argument of the power conjunction (2 or 3) from the array that is the argument to the resulting derived verb. To see the real power of the power conjunction, consider that it accepts negative arguments as well:
+~(^:_1) 1 5 10 20 0.5 2.5 5 10
You have seen right: J can figure out that the inverse of adding a number to itself is dividing that number by two!
Pretty much any programming language permits you to assign values to names for re-use in later expressions. J is no exception:
data =. 1 5 10 20 double =. +~ double data 2 10 20 40 inv =. ^:_1 halve =. double inv halve data 0.5 2.5 5 10
As you can see, names can be given not just to nouns (i.e. data), but also to verbs, adverbs, and conjunctions. Most J programs are just pieces of expressions that are assigned to names. Which means that the short summary of J that I have given here could well be all you ever need to know about the language - apart from the fact that you will have to acquire a working knowledge of many more verbs, adverbs, and conjunctions.
Before you rush off to the Play Store looking for J, let me add that J is not yet there, although it's supposed to arrive soon. For now, you have to download the APK and install it yourself, using your preferred Android file manager. I should also point out that J is not just for Android. It's been around for more than 20 years, and you can get J for all the common computing platforms from Jsoftware. There's also an iOS version for the iPhone and iPad. J's extreme terseness is a perfect fit for smartphones, where screen space is a scarce resource and where every character you don't have to type saves you a lot of time.